De phakomatosis is een groep neurocutane aandoeningen van genetische oorsprong, zeldzaam bij de algemene bevolking. Op klinisch niveau worden ze gekenmerkt door de ontwikkeling van een multisystemische organische betrokkenheid bij huid- of tumorlaesies, in verschillende delen van de huid, organen of zenuwstelsel..
Bovendien maakt het niet-specifieke klinische verloop de vroege diagnose moeilijk, zodat de medische en psychologische gevolgen de kwaliteit van leven van de getroffen persoon en hun familieleden aanzienlijk verslechteren..
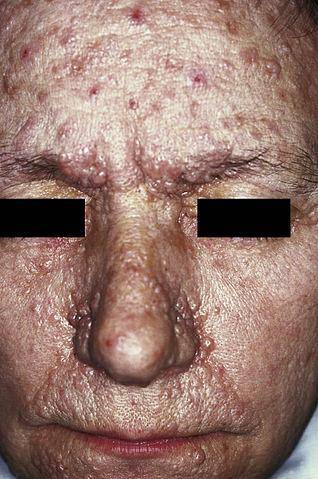
Hoewel er een groot aantal neurocutane ziekten is, zijn de meest voorkomende fibromatose type I en type II, de ziekte van Bourneville, het Sturge-Weber-syndroom en de ziekte van Von Hippel-Lindau..
Aan de andere kant, ondanks het feit dat dit allemaal aangeboren pathologieën zijn, zijn er meerdere therapeutische benaderingen van dermatologische aard ontworpen die proberen de tekenen en symptomen die kenmerkend zijn voor deze aandoeningen en daarmee de medische prognose van de getroffenen te verbeteren..
Artikel index
De term phakomatosis komt van de uitdrukking van Griekse oorsprong Phakos waarvan de betekenis verwijst naar <
Neurocutane pathologieën worden fundamenteel gekenmerkt door het bestaan van een significant verband tussen een neurologische aandoening of aandoening en de dermatologische manifestaties.
De term neurocutane pathologie wordt dus op een algemene manier gebruikt om verschillende ziekten te omvatten die aanwezig zijn bij de congenitaal aangedane persoon en die bovendien gedurende het hele leven aanwezig kunnen zijn met de ontwikkeling van huidlaesies en tumoren in verschillende gebieden, zenuwstelsel, cardiovasculair systeem, niersysteem, huidsysteem, oftalmisch systeem, enz..
Op deze manier werd de term phakomatosis in 1917 geïntroduceerd door Brouwer en later door van der Hoeve in 1923, maar de eerste beschrijvingen verwezen alleen naar enkele pathologieën die in deze groep waren opgenomen. Momenteel meer dan 40.
Op klinisch niveau wordt phakomatosis beschreven als een ziekte die zich presenteert met huidveranderingen en goedaardige / kwaadaardige misvormingen in verschillende systemen: neurologisch, oculair, cutaan en visceraal..
Met betrekking tot de getroffen gebieden wijzen verschillende auteurs erop dat die van ectodermale oorsprong het meest worden aangetast, dat wil zeggen de huid en het zenuwstelsel, hoewel ze ook andere systemen of apparaten, zoals het oog, kunnen aantasten..
Syndromen en pathologieën van neurocutane oorsprong zijn zeldzame ziekten bij de algemene bevolking, hoewel er op algemeen niveau geen specifieke gegevens over al deze ziekten zijn..
De epidemiologie van deze aandoeningen varieert dus afhankelijk van het type ziekte, in het bijzonder is neurofibromatose een van de meest voorkomende, met een relatieve prevalentie van één geval per 300.000 geboorten..
Neurocutane ziekten worden gekenmerkt door de ontwikkeling van huidlaesies. Specifiek onderscheidt phakomatosis zich van vele anderen door de aanwezigheid van hamartomen..
Hamartomen zijn een soort goedaardige misvorming of tumor die kan groeien in verschillende organen, zoals de hersenen, het hart, de ogen, de huid of de longen..
Phakomatosis kan echter worden geassocieerd met een groot aantal medische aandoeningen die fundamenteel zullen variëren, afhankelijk van de specifieke ziekte of pathologie waaraan de getroffen persoon lijdt..
Momenteel is een groot aantal neurocutane aandoeningen vastgesteld op klinisch en genetisch niveau, maar er zijn er enkele met een hogere prevalentie in de algemene bevolking: neurofibromatose type I en type II, de ziekte van Bourneville, de ziekte van Von Hippel-Lindau en Sturge-Weber syndroom.
Er zijn verschillende klinische vormen van neurofibromatose. Momenteel zijn de meest voorkomende echter type I neurofibromatose, ook wel de ziekte van Von Reclinghausen genoemd, en type II neurofibromatose, gevolgd door spinale shwannomatose..
Op etiologisch niveau hebben al deze medische manifestaties van neurofibromatose een genetische oorsprong en komen ze voor bij de vorming van tumoren in zenuwgebieden, vooral het centrale en perifere zenuwstelsel..
Tumorformaties, meestal niet-kankerachtig of goedaardig, hebben de neiging bijna overal in het zenuwstelsel te groeien en zich te ontwikkelen, zoals de hersenen, het ruggenmerg of de perifere zenuwen.
Algen met secundaire medische complicaties bij neurofibromatose omvatten dus onder meer afwijkingen in de groei, de ontwikkeling van aanvallen, het optreden van hersentumoren, botpathologieën, doofheid en / of blindheid of de ontwikkeling van belangrijke leerproblemen..
Bovendien is deze pathologie aanwezig vanaf het moment van geboorte. De significante manifestatie van het klinische beeld kan echter worden uitgesteld tot de late kindertijd, vroege adolescentie of volwassenheid..
Aan de andere kant omvat de diagnose van dit type pathologie meestal, naast het lichamelijk en neurologisch onderzoek, verschillende neuroimaging-tests en genetische analyses..
Bovendien is er momenteel geen remedie voor neurofibromatose, maar er zijn gespecialiseerde therapeutische benaderingen voor het beheersen van dermatologische aandoeningen, deze kunnen zowel farmacologische als chirurgische behandelingen omvatten om tumorvorming te stoppen of te elimineren..
Neurofibromatose type I (NF1), ook bekend als de ziekte van von Recklinghausen, manifesteert zich voornamelijk door de aanwezigheid van lichtbruine vlekken, gewoonlijk 'café au lait' genoemd, ephelides (sproeten) en neurofibromen (zenuwbeschadiging in Schwann-cellen en neurieten).
Het heeft een autosomaal dominante genetische oorsprong, met name door een mutatie op chromosoom 17, op locatie 17q11.2. Dus het gen dat betrokken is bij
de ontwikkeling van type I neurofibromatose, speelt een prominente rol bij de modulatie van celgroei en differentiatie en kan daarnaast functioneren als tumoronderdrukker.
Met betrekking tot de epidemiologie van deze pathologie, presenteert het een geschatte prevalentie van één geval voor elke 2.500.3000 geboorten.
De diagnose van type I neurofibromatose wordt meestal gesteld op basis van de consensus klinische criteria van het National Institute of Health (1987), maar vereist continue follow-up om secundaire medische complicaties te voorkomen..
Normaal gesproken worden tumorgroei behandeld met medicijnen om hun exponentiële ontwikkeling te voorkomen of door chirurgische verwijdering.
Neurofibromatose type II (NF2), manifesteert zich voornamelijk door de ontwikkeling van schwannomen, dat wil zeggen tumorformaties afgeleid van Shcwaan-cellen die verantwoordelijk zijn voor het bedekken van de zenuwverlengingen.
Schwannomen of neuriomen tasten meestal vooral de gehoor- en oogzenuw aan en in mindere mate de huidgebieden.
Type II neurofibromatose heeft een autosomaal dominante genetische oorsprong, met name door de aanwezigheid van een mutatie op chromosoom 22, op locatie 22q11.22.
Het gen dat betrokken is bij de ontwikkeling van deze pathologie is verantwoordelijk voor het coderen van een eiwitcomponent met een prominente rol bij de onderdrukking van tumoren, dus de gebrekkige activiteit ervan veroorzaakt een abnormale toename van celproliferatie.
Met betrekking tot de epidemiologie van deze pathologie, komt deze minder vaak voor dan type 1, met een geschatte prevalentie van één geval per 50.000 geboorten.
De diagnose van type II neurofibromatose is vergelijkbaar met het vorige type en wordt meestal gesteld op basis van de consensus klinische criteria van het National Institute of Health. Het omvat echter meestal aanvullende laboratoriumtests, zoals neuroimaging..
Normaal worden tumorgroei met medicijnen behandeld, maar in gevallen waarin dit mogelijk is, wordt chirurgische verwijdering gebruikt.
De ziekte van Bourneville is een van de termen die worden gebruikt om te verwijzen naar tubereuze sclerose, een genetische aandoening die wordt gekenmerkt door de aanwezigheid van hamartomen.
Op klinisch niveau kan het aanleiding geven tot een multisystemische aandoening die wordt gekenmerkt door huidaandoeningen (angiomen in het gezicht, nagelfibromen, fibreuze plaques, hypochrome vlekken, enz.), Nieraandoeningen (renale angiomyolipomen of niercysten), hartaandoeningen (cardiale rabdomyomen) neurologische aandoening (corticale knollen, subependymale gliale knobbeltjes, atrocytomen, toevallen, verstandelijke beperking, gedrags- en motorische afwijkingen), onder andere.
Net als de hierboven beschreven ziekten, is de oorsprong van tubereuze sclerose genetisch bepaald. Specifiek is het te wijten aan de aanwezigheid van mutaties in de TSC1- en TSC2-genen..
Anderzijds wordt de diagnose tubereuze sclerose gesteld op basis van de klinische criteria die zijn voorgesteld op een medische conferentie in 1998. De genetische studie wordt echter ook relevant geacht voor de bevestiging ervan..
Met betrekking tot de behandeling van tubereuze sclerose, hoewel er geen genezing is, worden meestal verschillende farmacologische en chirurgische benaderingen gebruikt, voornamelijk voor de beheersing van tumorgroei en secundaire medische complicaties zoals neurologische manifestaties..
De ziekte van Von Hippel-Lindau, ook bekend als retino-cerebellaire angiomatose, manifesteert zich voornamelijk door de aanwezigheid en ontwikkeling van vasculaire misvormingen, cysten en / of tumoren, meestal goedaardig..
Het heeft een autosomaal dominante genetische oorsprong, met name door een mutatie op chromosoom 3, op locatie 3p-25-26. Bovendien presenteert het een geschatte incidentie van één geval per 40.000 geboorten..
In het bijzonder treft de ziekte van Von Hippel-Lindau voornamelijk het centrale zenuwstelsel (CZS) en het netvlies, door de vorming van hemangiomen.
Hemangiomen zijn vasculaire misvormingen die worden gekenmerkt door de aanwezigheid van clusters van verwijde bloedcapillairen. Ze verschijnen meestal in hersen- en ruggengraatgebieden, hoewel ze ook vaak voorkomen in het netvlies of op de huid.
De diagnose van deze pathologie vereist, naast het lichamelijk en neurologisch onderzoek, een gedetailleerd oftalmologisch onderzoek, samen met de analyse van verschillende neuroimaging-tests, om de aanwezigheid van zenuwlaesies te bevestigen..
Wat betreft de behandeling van de ziekte van Von Hippel-Lindau, is de basisinterventie een operatie om vasculaire misvormingen te elimineren. Het vereist echter continue monitoring om secundaire complicaties te voorkomen..
Bovendien heeft het een verminderde levensverwachting, rond de leeftijd van 50 jaar, voornamelijk als gevolg van de ontwikkeling van niercelcarcinomen (neoplastische formaties van kankercellen in de niertubuli).
Sturge-Weber-syndroom, ook bekend als encefalo-trigeminale angiomatose, manifesteert zich voornamelijk door de aanwezigheid van hemangiomen.
Een hemangioom is een type neoplasma of tumorvorming dat wordt gekenmerkt door de aanwezigheid van een abnormaal hoog aantal bloedvaten in de huid of andere inwendige organen..
Specifiek, op klinisch niveau, wordt het Sturge-Weber-syndroom gekenmerkt door de ontwikkeling van hemangiomen in het gezicht, intracraniële hemangiomen en choridische, conjunctivale, episcerale en glaucoomhemangiomen..
Het heeft een genetische oorsprong, met name door een mutatie op chromosoom 9, op locatie 9q21, in het GNQ-gen. Deze genetische component speelt een prominente rol bij de controle van groeifactoren, vasoactieve peptiden en neurotransmitters (Orhphanet, 2014).
De diagnose van het Sturge-Weber-syndroom wordt gesteld op basis van klinische verdenkingen en verschillende laboratoriumtests, zoals computertomografie of magnetische resonantiebeeldvorming..
Anderzijds kan lasertherapie in termen van behandeling de progressie van deze pathologie verminderen en bovendien in veel gevallen hemangiomen volledig elimineren..



Niemand heeft nog op dit artikel gereageerd.