
De Prader-Willi-syndroom SPW) is een multisystemische pathologie met een aangeboren genetische oorsprong. Het is een complexe ziekte die eetlust, groei, metabolisme, gedrag en / of cognitieve functie beïnvloedt.
Op klinisch niveau, tijdens de kindertijd, wordt deze ziekte gekenmerkt door de aanwezigheid van verschillende medische bevindingen zoals spierzwakte, eetstoornissen of gegeneraliseerde ontwikkelingsachterstand..
Bovendien vertoont een groot deel van de personen die getroffen zijn door het Prader-Willi-syndroom op cognitief en gedragsniveau een matige verstandelijke beperking of vertraging die gepaard gaat met verschillende leer- en gedragsproblemen..
Hoewel het Prader-Willi-syndroom als een zeldzame of ongebruikelijke ziekte wordt beschouwd, geven talrijke onderzoeken aan dat het een van de meest voorkomende pathologieën op genetisch gebied is. De diagnose van deze ziekte wordt voornamelijk gesteld op basis van klinische bevindingen en complementaire genetische tests..
Met betrekking tot de behandeling is er nog geen remedie voor het Prader-Willi-syndroom, dus de therapeutische benadering is gericht op het behandelen van de symptomen en complicaties, waarbij obesitas de medische bevinding is die de grootste bedreiging vormt voor de getroffenen..
Dus, met betrekking tot de prognose en kwaliteit van leven, zullen beide afhangen van de ernst van de bijbehorende medische problemen en de gedrags- of cognitieve stoornissen die zich kunnen ontwikkelen..
Artikel index
Verschillende klinische rapporten geven aan dat het Prader-Willi-syndroom (PWS) aanvankelijk werd beschreven door J. L. Down, in 1887, na de diagnose 'polysarcia' bij een van zijn patiënten..
Het waren echter Drs Prader, Labhart en Willi die in 1956 nog eens 9 gevallen beschreven en deze pathologie zijn naam gaven. Bovendien werden de kenmerken en diagnostische criteria van het Prader-Willi-syndroom gesystematiseerd door Holm et al..
Prader-Willi-syndroom is een aangeboren genetische verandering, dat wil zeggen, het is een pathologie die aanwezig is vanaf het moment van geboorte en die het individu zijn hele leven zal beïnvloeden als er geen genezende therapeutische interventie is..
Deze pathologie vertoont een complex klinisch beloop, gekenmerkt door talrijke medische manifestaties.
Hoewel het fenotype van het Prader-Willi-syndroom tegenwoordig nauwkeuriger bekend is, is er in de afgelopen 25 jaar aanzienlijke vooruitgang geboekt bij de analyse en het begrip van deze ziekte..
De uitdrukking van het Prader-Willis-syndroom is divers, het heeft de neiging om meerdere systemen en structuren te beïnvloeden, waarbij de meeste veranderingen verband houden met hypothalamische disfunctie.
De hypothalamus is een neurologische structuur die een essentiële rol speelt bij de controle van homeostatische functies: de regulering van honger, dorst, slaap-waakcycli of de regulering van de lichaamstemperatuur.
Bovendien geeft de hypothalamus verschillende hormonen af aan verschillende klieren: groei, seksueel, schildklier, enz..
Ten slotte moeten we erop wijzen dat er in de medische en experimentele literatuur ook naar Prader-Willis-syndroom kan worden verwezen met andere termen, zoals het Prader-Labhart-Willi-syndroom of met het acroniem PWS..
Andere synoniemen zijn ook het Labhart Willi-syndroom, het Praser Labhart Willi Fancone-syndroom of het hypogenitale dystrofiesyndroom..
Prader-Willi-syndroom (PWS) is een zeldzame genetische ziekte. De term zeldzame ziekte (ER) wordt gebruikt om te verwijzen naar die pathologieën die zeldzaam zijn of weinig mensen die eraan lijden.
Momenteel wordt geschat dat het Prader-Willi-syndroom een pathologie is met een geschatte frequentie van 1 geval per 10.000-30.000 mensen wereldwijd.
Aan de andere kant, met betrekking tot de verdeling naar geslacht, is opgemerkt dat deze pathologie mannen en vrouwen in gelijke mate treft en niet wordt geassocieerd met etnische groepen of geografische regio's..
Bovendien wordt het Prader-Willi-syndroom beschouwd als de belangrijkste oorzaak van obesitas van genetische oorsprong.
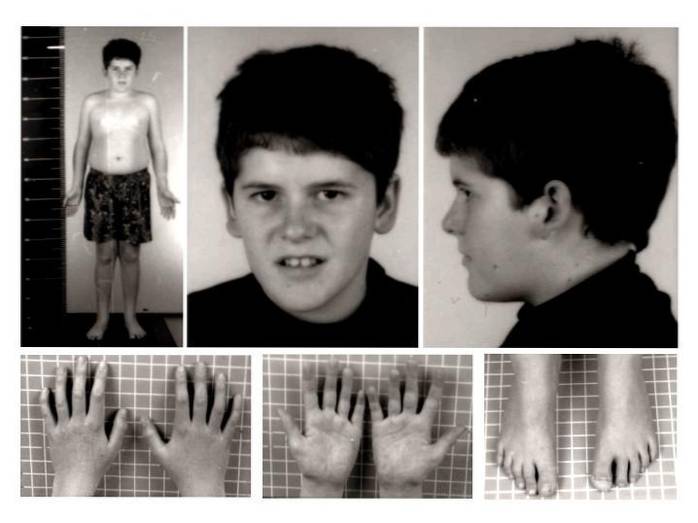
Op klinisch niveau wordt het Prader-Willi-syndroom traditioneel in verband gebracht met neonatale hypotonie, hypogonadisme, hyperfagie, zwaarlijvigheid, kleine gestalte, gegeneraliseerde ontwikkelingsachterstand, matige verstandelijke beperking, atypische gezichtsuitdrukkingen en verschillende gedragsveranderingen..
Desondanks is de klinische expressie van deze pathologie zeer heterogeen en varieert aanzienlijk tussen de getroffen individuen..
Bovendien variëren de karakteristieke tekenen en symptomen van het Prader-Willi-syndroom afhankelijk van de biologische ontwikkeling, zodat we verschillende klinische bevindingen kunnen waarnemen in de foetale en neonatale periode, de periode van de kindertijd of vroege kinderjaren, de schoolfase en tenslotte de fase Tiener.
Op een systematische manier beschrijven José A. del Barrio del Campo en medewerkers in detail de meest karakteristieke veranderingen op biomedisch, psychomotorisch, cognitief en gedragsgebied:
De meest kenmerkende fysieke tekenen en symptomen zijn onder meer stoornissen zoals; hypotonie, musculoskeletale misvormingen of misvormingen, verminderd of laag gewicht en lengte, overmatige eetlust, zwaarlijvigheid, hypogonadisme, slaapstoornissen, ademhalingsstoornissen, atypische gemakkelijke eigenschappen, verandering in de regulering van de lichaamstemperatuur, onder andere.
Aanwezigheid of ontwikkeling van verminderde spierspanning. De spierverslapping bij deze pathologie wordt vooral geaccentueerd in de nek en romp, vooral in de neonatale fase en de eerste levensmaanden. Dus met biologische ontwikkeling neigt de spiertonus te verbeteren.
In dit geval is het gebruikelijk om de ontwikkeling van scoliose of afwijking van de wervelkolom, een slechte uitlijning van de onderste ledematen (genu valgus) of de aanwezigheid van platvoeten waar te nemen..
Daarnaast kunnen ook andere soorten aangeboren afwijkingen worden waargenomen, zoals verkleining van de voet en handen, heupdysplasie, aanwezigheid van zes vingers, onder anderen..
Vooral bij de geboorte zijn zowel de lengte als het gewicht van het getroffen kind lager dan verwacht voor hun ontwikkeling en geslacht. Ondanks het feit dat standaardwaarden kunnen worden bereikt op volwassen leeftijd, heeft de langzame groeisnelheid de neiging om volwassen waarden van lengte en gewicht te veranderen.
Het is gebruikelijk om bij mensen met het Prader-Willi-syndroom een onverzadigbare eetlust waar te nemen, gekenmerkt door een obsessie of fixatie op voedsel. Door de inname van grote hoeveelheden voedsel hebben de getroffenen de neiging om zwaarlijvigheid en andere daarmee samenhangende medische complicaties te ontwikkelen, zoals diabetes mellitus type II..
De aanwezigheid van genitale veranderingen komt ook vaak voor. Specifiek komt hypogonadisme of gedeeltelijke ontwikkeling van de uitwendige geslachtsorganen zeer vaak voor. In de meeste gevallen bereikt de puberale ontwikkeling de laatste of volwassen stadia niet.
Snurken, verhoogde frequentie of ademstilstand komen vaak herhaaldelijk voor tijdens de slaapfasen. De getroffenen hebben dus de neiging om verschillende veranderingen te vertonen die verband houden met fragmentatie, slaapvertraging of de aanwezigheid van periodiek ontwaken.
Afwijkingen en misvormingen van het bewegingsapparaat kunnen ook craniofaciale kenmerken beïnvloeden. Het is mogelijk om een smalle schedel, oogscheelzien, slecht gepigmenteerde huid en haar, kleine mond en dunne lippen, tandmisvormingen, enz..
Mensen met het Prader-Willi-syndroom hebben meestal problemen die verband houden met de regulering van de lichaamstemperatuur, en een andere belangrijke bevinding is een hoge pijnbestendigheid.
Door de aanwezigheid van musculoskeletale misvormingen en verminderde spierspanning, zal de psychomotorische ontwikkeling langzamer verlopen en alle gebieden aantasten.
De getroffenen hebben meestal serieproblemen bij het uitvoeren van elk type activiteit dat een of meer motorische uitvoeringen vereist.
Wat betreft cognitieve beperkingen: de meeste getroffenen hebben een lichte of matige verstandelijke beperking.
Daarnaast presenteren ze meestal enkele specifieke gebieden die meer worden beïnvloed, zoals sequentiële verwerking van informatie, recent of kortetermijngeheugen, oplossen van rekenkundige problemen, auditieve verwerking van verbale informatie, verandering van aandacht en concentratie en aanwezigheid van cognitieve rigiditeit.
Aan de andere kant is taal een ander gebied dat aanzienlijk wordt beïnvloed bij personen met het Prader-Willi-syndroom. Vertragingen bij het verwerven van fonologische vaardigheden, slechte woordenschat, wijziging van grammaticale constructie, onder andere worden gewoonlijk waargenomen..
Gedragsproblemen en -veranderingen zijn een van de typische bevindingen die kunnen worden waargenomen bij het Prader-Willi-syndroom, ze variëren normaal gesproken afhankelijk van de leeftijd of het rijpingsstadium waarin de getroffen persoon zich bevindt, maar enkele van de meest voorkomende gedragskenmerken zijn:
Diverse lopende onderzoeken hebben aangetoond dat gedragsverandering de neiging heeft toe te nemen met de leeftijd en daarom de neiging heeft om te verslechteren, waarbij ze op een algemene manier sociale, familiale en emotionele gebieden aantasten..
Zoals we in verschillende secties hierboven hebben opgemerkt, heeft het Prader-Willi-syndroom een genetische oorsprong.
Hoewel er momenteel een grote controverse bestaat over de specifieke genen die verantwoordelijk zijn voor deze pathologie, tonen alle gegevens aan dat de etiologische verandering zich op chromosoom 15 bevindt..
Tijdens de genetische studie van deze pathologie zijn er verschillende bijdragen geweest. Burtler en Palmer (1838) ontdekten de aanwezigheid van afwijkingen in de lange arm van chromosoom 15 van de vaderlijke ouder, terwijl Nicholls (1989) opmerkte dat in andere gevallen de aandoening verband hield met chromosomale veranderingen van de moeder (Rosell-Raga, 2003).
Afgezien hiervan is de meest geaccepteerde theorie over de oorsprong van deze pathologie het verlies of de inactivering van verschillende genen van vaderlijke expressie die zich in het 15q11-13-gebied van chromosoom 15 bevinden..
De diagnose van het Prader-Willi-syndroom heeft twee basiscomponenten: de analyse van klinische bevindingen en genetische tests..
Wat betreft de detectie van indicatortekens en symptomen, zowel bij baby's als bij oudere kinderen, is het essentieel om een gedetailleerde, individuele en familiale medische geschiedenis op te stellen. Evenzo is het ook essentieel om een lichamelijk en neurologisch onderzoek uit te voeren..
Als er op basis van deze procedures een diagnostisch vermoeden bestaat, zal het nodig zijn om verschillende aanvullende tests voor te schrijven om de aanwezigheid van genetische veranderingen en afwijkingen vast te stellen..
Concreet wordt ongeveer 90% van de gevallen definitief gediagnosticeerd door middel van DNA-methylatietests en andere aanvullende tests..
Bovendien is het ook mogelijk om een prenatale diagnose van deze medische aandoening te stellen, voornamelijk in gezinnen met een voorgeschiedenis van het Prader-Willi-syndroom..
Concreet maakt de vruchtwaterpunctie de extractie van embryostalen mogelijk voor de uitvoering van de relevante genetische tests.
Er is momenteel geen remedie voor het Prader-Willi-syndroom. Net als bij andere zeldzame ziekten zijn de behandelingen beperkt tot symptoomcontrole en verbetering van de kwaliteit van leven van de getroffen mensen.
Een van de fundamentele aspecten zal echter voeding en dieetcontrole zijn, aangezien obesitas de belangrijkste oorzaak is van morbiditeit en mortaliteit bij deze pathologie..
Aan de andere kant vereist de aanwezigheid van cognitieve en gedragsveranderingen de tussenkomst van gespecialiseerde professionals, zowel bij cognitieve revalidatie als bij de behandeling van gedragsstoornissen..



Niemand heeft nog op dit artikel gereageerd.