De Duchenne spierdystrofie (DMD) is een neuromusculaire aandoening, gekenmerkt door de aanwezigheid van aanzienlijke spierzwakte en een gegeneraliseerde en progressieve ontwikkeling (Wereldgezondheidsorganisatie, 2012).
Het is het meest voorkomende type spierdystrofie bij mensen (López-Hernández, 2009) en treft 1 op de 3.500 kinderen in de wereld (Duchenne Parent Project, 2012). De ziekte treft voor het grootste deel mannen in de vroege levensfasen (Wereldgezondheidsorganisatie, 2012).
Er zijn verschillende soorten spierdystrofie. Symptomen beginnen meestal tijdens de kindertijd. Zwakte en verlies van spiermassa veroorzaken ernstige moeilijkheden bij het verwerven of behouden van het vermogen om te lopen, ademen en / of slikken (Mayo Clinic, 2013).
Neuromusculaire effecten bieden een chronische prognose. In de meeste gevallen sterven mensen met spierdystrofie van Duchenne op jonge leeftijd als gevolg van de ontwikkeling van secundaire pathologieën zoals hartfalen of cardiomyopathieën (Wereldgezondheidsorganisatie, 2012).
Artikel index
Duchenne-spierdystrofie is een ziekte die het individu treft door progressieve spierzwakte en degeneratie (Muscular Dystrophy Association, 2016).
Door een genetische mutatie zal de afwezigheid van een specifiek eiwit bij mensen met spierdystrofie van Duchenne leiden tot verlies van spierfunctionaliteit.
Over het algemeen verschijnen de symptomen meestal in de onderste ledematen en verspreiden ze zich naar de rest van de gebieden.
De Wereldgezondheidsorganisatie (2012) geeft aan dat de incidentie van spierdystrofie van Duchenne wordt geschat op ongeveer 1 geval per 3.300 inwoners.
In het bijzonder toont enig onderzoek aan dat deze ziekte 1 op de 3.500 levend geboren mannelijke kinderen treft (López-Hernández, 2009).
In het geval van de VS is het niet met zekerheid bekend hoeveel mensen in alle leeftijdscategorieën aan deze ziekte lijden. Sommige onderzoeken hebben geschat dat één op de 5.600-7.770 mannelijke volwassenen in de leeftijd van 5 tot 24 jaar de diagnose Duchenne- of Becker-spierdystrofie heeft (Centers for Disease Control and Prevention, 2015).
Het meest kenmerkende van de aandoeningen die tot de groep van spierdystrofieën behoren, is de spierzwakte; Afhankelijk van het type kunnen er echter specifieke symptomen optreden die variëren afhankelijk van de aanvangsleeftijd en de getroffen spiergroepen (Mayo Clinic, 2013).
Normaal gesproken is de ontwikkeling van spierdystrofie van Duchnne vrij voorspelbaar. Ouders kunnen enkele vrij significante symptomen waarnemen, zoals moeilijkheid of onvermogen om te leren lopen of abnormale toename van kuitspieren (pseudohypertrofie) (Duchenne Ouderproject, 2012).
Enkele van de meest kenmerkende symptomen en tekenen van spierdystrofie van Duchenne die vroeg in het leven van een kind optreden, zijn (Mayo Clinic, 2013):
Evenzo belicht de vereniging Duchenne Parent Projet (2012) de meest voorkomende symptomen en klinische manifestaties:
Alle spiersymptomen beginnen met zwakte van de spieren van de bekkengordel, kuiten en verschillende gangstoornissen die significant zijn vóór de leeftijd van 5 jaar (López-Hernández, 2009).
Op de kleuterschool kunnen kinderen met spierdystrofie van Duchenne vaak vallen of moeite hebben met lopen, trappen oplopen en / of rennen (Duchenne Parent Project, 2012).
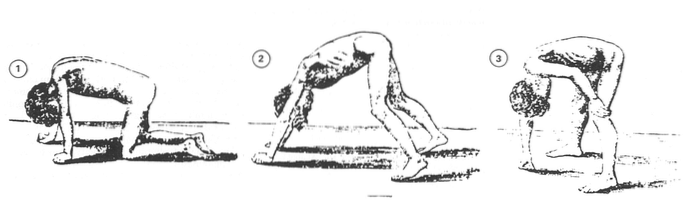
Naarmate de ziekte vordert, is het vrij waarschijnlijk dat kinderen op schoolleeftijd alleen de toppen van hun voeten gebruiken om te lopen. We zullen een rollende en onveilige mars kunnen observeren die talloze valpartijen kan veroorzaken. Ze gebruiken meestal een aantal strategieën om hun evenwicht te bewaren, zoals hun schouders naar achteren duwen of hun eigen lichaam vasthouden (Duchenne Parent Project, 2012).
Rond de leeftijd van 9 jaar kunnen de meeste mensen met deze ziekte niet lopen, hierdoor beginnen ze talrijke musculoskeletale misvormingen te ontwikkelen - scoliose, contracturen, enz. - (López-Hernández, 2009).
In de adolescente fase zullen ze aanzienlijke moeilijkheden ondervinden bij het efficiënt uitvoeren van activiteiten die verband houden met het gebruik van de bovenste ledematen, benen of romp. In dit stadium hebben ze mechanische ondersteuning en hulp nodig (Duchenne Parent Project, 2012).
Spierdegeneratie en spierzwakte gaan door totdat ze de spieren bereiken die verantwoordelijk zijn voor de ademhalings- en hartfunctie (López-Hernández, 2009). Door dit alles wordt het voortbestaan van de patiënt ernstig in gevaar gebracht, met in de meeste gevallen de dood tot gevolg..
Er zijn verschillende genen geïdentificeerd die betrokken zijn bij de productie van eiwitten die verantwoordelijk zijn voor het beschermen van spiervezels tegen mogelijke schade en letsel (Mayo Clinic, 2013).
In het bijzonder treedt elk type spierdystrofie op als gevolg van een bepaalde genetische mutatie. Sommige van deze mutaties zijn erfelijk; in de meeste gevallen treden ze echter spontaan op tijdens de zwangerschap (Mayo Clinic, 2013).
In het geval van spierdystrofie van Duchenne identificeerden de onderzoekers een specifiek gen op het X-chromosoom dat de mutatie zou kunnen presenteren die verantwoordelijk is voor deze pathologie (Muscular Dystrophy Association, 2016).
Zo werd in 1987 het eiwit dat met dit gen is geassocieerd, geïdentificeerd., dystrofine. Het ontbreken of ontbreken van dit eiwit impliceert dus dat de spieren kwetsbaar en gemakkelijk beschadigd zijn (Muscular Dystrophy Association, 2016).
Bovendien is een recessief overervingspatroon geïdentificeerd dat is gekoppeld aan het X-chromosoom, waarbij de drager de moeder is (Muscular Dystrophy Association, 2016). Vanwege dit feit komt dit type ziekte vaker voor bij mannen dan bij vrouwen..
Mannen hebben een XY-chromosoomsamenstelling, terwijl vrouwen XX zijn. Daarom, als een X-chromosoom een mutatie heeft in het DMD-gen, zal het lijden aan spierdystrofie van Duchenne vanwege de afwezigheid van dystrofineproductie (National Human Genome Research Institute, 2013).
In het geval van vrouwen die twee X-chromosomen hebben en dus twee kopieën van het DMD-gen, zal, als een van deze wordt gewijzigd, de andere in staat zijn om dystrofine te produceren en daardoor spierneuroprotectie te behouden (National Human Genome Research Institute, 2013 ).
Bij dit soort pathologieën kunnen verschillende interventies worden uitgevoerd om de diagnose te bepalen (National Human Genome Research Institute, 2013).
De klinische diagnose kan al worden gesteld wanneer een kind progressieve spierzwakte begint te ontwikkelen. Al op 5-jarige leeftijd zijn er duidelijke symptomen. Als er geen vroege interventie wordt uitgevoerd, zullen kinderen vóór de leeftijd van 13 jaar functionele afhankelijkheid vertonen (National Human Genome Research Institute, 2013).
Naast observatie en klinisch onderzoek kunnen enkele van de volgende technieken worden gebruikt om de aanwezigheid van Duchenne-spierdystrofie te identificeren (Mayo Clinic, 2013):
Op dit moment is er geen remedie voor spierdystrofie van Duchenne geïdentificeerd (Duchenne Parent Project, 2012).
Desondanks worden er verschillende behandelingen gebruikt waarvan is bewezen dat ze effectief zijn, zowel voor het verminderen van symptomen als voor het verbeteren van de kwaliteit van leven van mensen die aan dit soort pathologie lijden (Duchenne Parent Project, 2012).
Deze ziekte vereist, vanwege de klinische progressie en de grote verscheidenheid aan symptomen, een multidisciplinaire en uitgebreide interventie die wordt uitgevoerd door een breed scala aan specialisten: kinderarts, fysiotherapeut, neuroloog, neuropsycholoog, ergotherapeut, logopedist, voedingsdeskundige, endocrinoloog, geneticus, o.a. cardioloog, longarts, orthopedist, revalidator en chirurg (Duchenne Parent Project, 2012).
In veel gevallen kunnen specialisten farmacologische interventies aanbevelen (Mayo Clinic, 2013):
Geneesmiddelen zijn niet alleen nuttig voor interventie bij spierdystrofie van Duchenne, er zijn zowel therapeutische interventies als zorgmethoden die de kwaliteit van leven van deze mensen kunnen verbeteren (Mayo Clinic, 2013).
Enkele nuttige interventies zijn (Duchenne Parent Project, 2012):
Tot relatief weinig jaren geleden overleefden mensen met spierdystrofie van Duchenne niet veel langer na het bereiken van de adolescentie (Muscular Dystrophy Association, 2016).
De grote vorderingen in medisch, technisch en genetisch onderzoek hebben zowel de progressie van de ziekte kunnen vertragen als een aanzienlijke verhoging van de kwaliteit van leven van mensen die eraan lijden (Muscular Dystrophy Association, 2016). Op deze manier is hart- en ademhalingszorg essentieel voor het behoud van vitale functies (Muscular Distrophy Association, 2016).
In veel gevallen zijn ze in staat de post-adolescente stadia te bereiken. Er worden steeds meer gevallen van spierdystrofie van Duchenne beschreven bij volwassenen van in de 30, inclusief mensen die in de leeftijd van 40 en 50 overleven (Muscular Dystrophy Associatin, 2016).
Momenteel zijn klinische proeven en onderzoek gericht op de ontwikkeling van gentherapieën die mutaties en tekortkomingen in de productie van dystrofine modificeren (Muscular Dystrophy Association, 2016).
Enkele van de meest onderzochte mechanismen zijn (López-Hernández, 2009):
Duchenne spierdystrofie is een ernstig invaliderende ziekte bij zowel kinderen als jonge volwassenen met een verwoestende prognose..
Ondanks het feit dat klinisch en experimenteel onderzoek belangrijke vorderingen heeft gemaakt bij de behandeling van symptomen, is er nog steeds geen remedie voor dit type pathologie..
Een grondig begrip van de biologische en genetische basis is essentieel om een genezende behandeling voor spierdystrofie van Duchenne te vinden..

Niemand heeft nog op dit artikel gereageerd.