De Canavan-ziekte Het is een zeldzame genetische ziekte die optreedt omdat zenuwcellen in de hersenen beschadigd zijn en niet met elkaar kunnen communiceren. Deze ziekte komt voor in elke samenleving en etnische groep, hoewel het veel vaker voorkomt bij de Ashkenazi-joodse bevolking en hun nakomelingen, waar 1 op de 6.400-13.000 mensen wordt getroffen. De wereldwijde prevalentie is onbekend.
Deze ziekte behoort tot de groep van leukodystrofieën. Deze categorie omvat alle genetische aandoeningen waarbij de myeline-omhulling die de axonen van neuronen omgeeft, is beschadigd en er daarom geen goede communicatie is tussen neuronen..
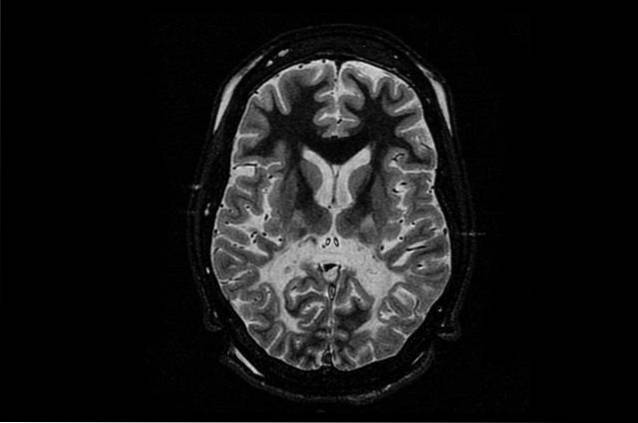
De meest voorkomende en tegelijkertijd meest ernstige vorm van deze ziekte is neonataal of infantiel. Deze vorm van de ziekte van Canavan treft pasgeboren kinderen of in hun eerste levensjaren..
Kinderen die aan deze ziekte lijden, hebben de eerste levensmaanden geen problemen, maar deze beginnen tussen 3 en 5 maanden te bloeien. De belangrijkste symptomen zijn te wijten aan het achterstand in ontwikkeling, waarbij kinderen motorische problemen hebben waardoor ze zich niet kunnen omdraaien, hun hoofd draaien of zonder enige ondersteuning zitten.
Andere veel voorkomende symptomen zijn spierzwakte (hypotonie), abnormale ontwikkeling van het hoofd (macrocefalie) en prikkelbaarheid. In mindere mate kunnen ze ook problemen hebben met eten, toevallen en slaapproblemen..
Een andere, minder vaak voorkomende vorm is de ziekte van Canavan die begint in de midden kinderjaren of adolescentie. Kinderen en adolescenten met deze ziekte hebben problemen met taalontwikkeling en motorische vaardigheden, maar deze problemen zijn vaak zo mild dat ze niet worden geïdentificeerd als symptomen van de ziekte van Canavan..
De levensverwachting van mensen met de ziekte van Canavan is zeer heterogeen en varieert met name naargelang het tijdstip waarop de ziekte begint..
Kinderen die aan de neonatale of infantiele vorm lijden, leven meestal maar een paar jaar, hoewel sommigen de adolescentie bereiken en zeer weinigen tot de volwassenheid. Terwijl degenen die lijden aan de juveniele vorm een normale levensverwachting hebben.
Artikel index
Er zijn twee goed gedifferentieerde vormen van de ziekte van Canavan: die met een neonataal of infantiel begin en die met aanvang in de midden kinderjaren of adolescentie..
Symptomen van de ziekte van Canavan bij pasgeborenen of bij kinderen zijn zeer ernstig, meestal niet merkbaar tot een leeftijd van 3-50 maanden, en omvatten macrocefalie, verlies van motorische controle over het hoofd en ontwikkelingsstoornissen. Ontwikkelingsachterstanden worden duidelijker naarmate het kind ouder wordt.
De ernstigste symptomen zijn die welke verband houden met motorische problemen, aangezien kinderen niet kunnen zitten of opstaan zonder ondersteuning, lopen of spreken. Als ze ouder worden, kan hypotonie tot spasticiteit leiden.
Hoewel ze al deze motorische problemen hebben, kunnen ze leren sociaal om te gaan, te glimlachen, naar objecten te wijzen ...
Sommige kinderen hebben ook last van optische atrofie, wat visuele problemen veroorzaakt, hoewel ze objecten nog steeds visueel kunnen identificeren.
Naarmate de symptomen toenemen, worden ze erger, waardoor ze slaapproblemen, toevallen en voedingsproblemen veroorzaken. Het kind wordt volledig afhankelijk en heeft hulp nodig bij het uitvoeren van elke taak.
De levensverwachting van deze kinderen is vrij kort, de meesten sterven binnen een paar jaar, hoewel sommigen leven tot de adolescentie of volwassenheid.
De ziekte van Canavan die midden in de kindertijd of adolescentie begint, is milder dan de vorige. Symptomen zijn onder meer enkele problemen bij verbale en motorische ontwikkeling.
Hoewel ze meestal zo mild zijn dat ze niet worden geïdentificeerd als symptomen van de ziekte van Canavan, wordt deze ziekte meestal gediagnosticeerd na het uitvoeren van een urineonderzoek, aangezien een van de markers de hoge concentratie van N-acetylasparaginezuur (NAA in de urine) is..
Deze ziekte wordt veroorzaakt door een mutatie in een gen genaamd ASPA. Dit gen regelt het enzym aspartoacylase, dat verantwoordelijk is voor het afbreken van NAA-moleculen..
De mutatie van het ASPA-gen zorgt ervoor dat aspartoacylase zijn effectiviteit vermindert, dus het zal niet genoeg NAA-moleculen afbreken en er zal een hoge concentratie van deze stof zijn. Hoe eerder deze mutatie optreedt, hoe slechter de effecten zijn.
Hoewel de werking van NAA-moleculen niet erg goed wordt begrepen, lijkt het erop dat ze betrokken zijn bij het transport van watermoleculen door neuronen en, de overmaat van deze stof, voorkomt dat nieuwe myeline wordt gevormd en vernietigt het bestaande. Hierdoor werken de verbindingen tussen neuronen niet goed en kunnen de hersenen zich niet normaal ontwikkelen..
Bovendien kan deze ziekte op autosomaal recessieve wijze worden overgeërfd. Dus als elk lid van het paar drager is van de pathogene variant van het ASPA-gen en ze besluiten een kind te krijgen, is de kans groot dat ze:
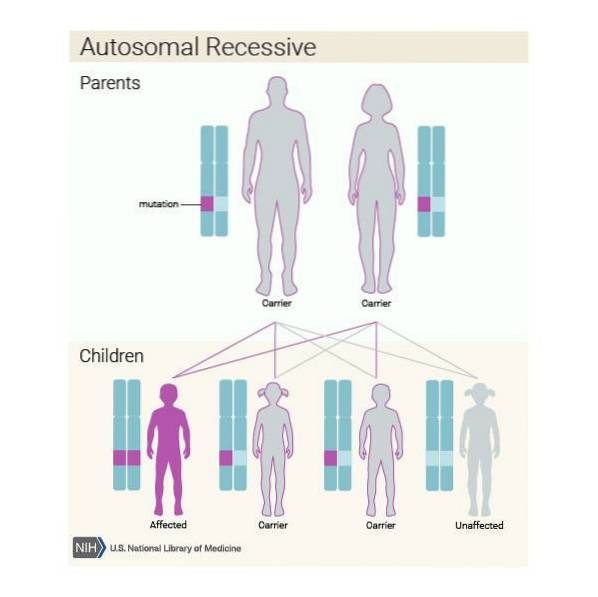
Het is erg belangrijk dat personen die tot de risicopopulatie behoren, in dit geval de afstammelingen van Asjkenazische joden, een genetische analyse ondergaan om te controleren of ze het ASPA-gen dragen voordat ze een kind krijgen..
De behandeling hangt af van de vorm van de ziekte en de symptomen die elk individu vertoont..
Er is momenteel geen remedie voor de ziekte van Canavan, dus beschikbare therapieën zijn gericht op het verbeteren van de kwaliteit van leven van de patiënt door ondersteuning, voeding en hydratatie, en het voorkomen en behandelen van infecties..
Het wordt aanbevolen dat kinderen een fysiotherapeutische behandeling krijgen om hun houding en motoriek te verbeteren, contracturen en spierproblemen, zoals decubitus, te voorkomen en te behandelen. Ze kunnen ook deelnemen aan therapeutische en educatieve programma's om hun communicatieve vaardigheden te verbeteren..
Behandeling met medicatie omvat anti-epileptica (AED's) als het kind epileptische aanvallen heeft, acetazolamide (merknaam Diamox) om de intracraniale druk en injecties met botulinumtoxine (Botox) om spasticiteit te behandelen, indien aanwezig.
Het is noodzakelijk om elke 6 maanden een follow-up uit te voeren om na te gaan in welke staat het kind verkeert en hoe het zich ontwikkelt.
Mensen die aan deze vorm van de ziekte lijden, ervaren veel mildere symptomen, dus hebben ze meestal alleen therapieën nodig om hun taal te verbeteren of speciale onderwijsprogramma's. Ze hebben geen medicatie nodig.
Jaarlijkse controle van de toestand van het kind wordt aanbevolen.
De werkzaamheid van andere therapieën wordt momenteel onderzocht bij zowel mensen als diermodellen..
De werkzaamheid van een genetische transplantatie in de hersenen van kinderen met de ziekte van Canavan wordt onderzocht met behulp van een niet-virale vector.
De eerste resultaten laten zien dat dit type transplantatie goed wordt verdragen door kinderen en enige biochemische, radiologische en metabolische veranderingen veroorzaakt, maar het is niet nuttig om de ziekte te genezen, dus worden er nog steeds tests uitgevoerd (Leone et al. 2000, Janson et al. . tot 2002).
McPhee et al. (2006) voeren een onderzoek uit waarin het gezonde ASPA-gen op verschillende plaatsen in het lichaam van kinderen wordt getransplanteerd, met AAV2 als vector. In een van de tests waaraan 10 vrijwillige kinderen deelnamen. Bij 3 van hen werkte de transplantatie en neutraliseerde ze hun antilichamen, maar geen van de kinderen verbeterde.
Lithiumcitraat kan de NAA-concentratie in de hersenen verlagen, daarom Assadi et al. (2010) besloten een experiment uit te voeren waarbij ze gedurende 60 dagen lithiumcitraat toedienden aan 6 mensen met de ziekte van Canavan.
NAA-concentratieniveaus in de basale ganglia en de witte stof van de frontale kwab werden gevonden, hoewel er geen klinische verbeteringen werden gevonden.
Het gebrek aan aspartoacylase-enzymen veroorzaakt lage acetaatspiegels in de hersenen, dus besloten Mahavarao en zijn team (2009) om glyceroltriacetaat te geven aan twee patiënten met de ziekte van Canaval om hun acetaatspiegel te verhogen en te kijken of dat ook toenam..
De verbinding werd goed verdragen door de patiënten, hoewel er geen klinische verbeteringen werden gevonden. Ze voeren momenteel tests uit waarbij een grotere hoeveelheid glyceroltriacetaat wordt toegediend.
Een van de manieren om diermodellen te maken die een ziekte vertegenwoordigen, is door dieren te maken knock out. Deze dieren, meestal muizen, zijn genetisch gemodificeerd om het gen dat bij de ziekte is veranderd te verwijderen of te veranderen. In dit geval is het gemodificeerde gen het ASPA-gen..
Diermodellen worden gebruikt om de ziekte beter te begrijpen, de biologische correlatie ervan te bestuderen en de doeltreffendheid van nieuwe behandelingen te verifiëren.
Matalon et al. (2003) gebruikte muizen knock out om de werkzaamheid van een gentherapie met AAV2 als vector te testen. Ze ontdekten dat er verbeteringen waren opgetreden in de myeline-omhulsels, maar alleen in sommige delen, niet in de hele hersenen.
Het team van Surendran heeft in samenwerking met de Genzyme Corporation (2004) een stamceltransplantatiebehandeling getest. Ze ontdekten dat er nieuwe oligodendrocyten waren geproduceerd, maar niet genoeg om alle myeline-omhulsels te herstellen..
Een ander team testte een therapie die bestond uit het vervangen van de defecte asparthoacyclase-enzymen door nieuwe die in het buikvlies van de muizen werden geïnjecteerd. knock out.
De resultaten op korte termijn toonden aan dat de enzymen de bloed-hersenbarrière konden passeren (hun doel bereikten) en de niveaus van NAA in de hersenen aanzienlijk konden verlagen. Hoewel deze resultaten veelbelovend zijn, is een longitudinale studie nodig om de langetermijneffecten te verifiëren (Zano et al., 2011).
De eerste tekenen die artsen erop wijzen dat er iets mis is, zijn de fysieke, vooral hypotonie en macrocefalie.
Als deze symptomen worden waargenomen, wordt normaal gesproken een neurouimaging-onderzoek bij het kind uitgevoerd om te controleren op tekenen van leukodystrofie, zoals een lagere dichtheid van witte stof. Opgemerkt moet worden dat deze test minder effectief is bij kinderen met de ziekte van Canavan die begint in de middelbare kinderjaren of adolescentie..
Zodra is bewezen dat het kind lijdt aan leukodystrofie, worden er meer specifieke tests gedaan om andere ziekten uit te sluiten, waaronder:
De laatste stap om de ziekte te bevestigen, zou zijn om als volgt een genetisch onderzoek uit te voeren:



Niemand heeft nog op dit artikel gereageerd.