
De Pallister-Killian-syndroom, ook bekend als tetrasomie 12, het is een zeldzame ziekte van genetische oorsprong die wordt gekenmerkt door een breed spectrum van betrokkenheid van meerdere organen.
Klinisch wordt deze pathologie gedefinieerd door verstandelijke beperking, psychomotorische achterstand, spierhypotonie, een atypisch gezichtsfenotype, pigmentafwijkingen in de huid en alopecia. Bovendien kunnen ook andere soorten medische complicaties optreden die verband houden met misvormingen in verschillende lichaamssystemen of epileptische aanvallen..
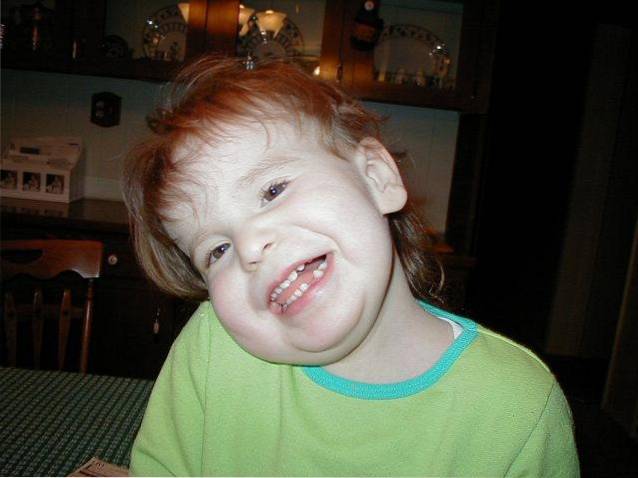
De etiologische oorsprong van deze ziekte is geassocieerd met een genetische aandoening die in mozaïek is verspreid. Specifiek is het te wijten aan de aanwezigheid van een extra chromosoom 12 in sommige cellen van het lichaam.
De diagnose Pallister-Killiam-syndroom kan zowel prenataal als postnataal worden gesteld. Het hoofddoel is de identificatie van de klinische kenmerken en het gebruik van een bevestigend genetisch onderzoek..
Dit syndroom heeft een hoog sterftecijfer. De farmacologische medische benadering en revalidatiebehandeling kunnen echter belangrijke voordelen opleveren voor de kwaliteit van leven en de klinische toestand van de getroffenen..
Artikel index
Deze ziekte werd aanvankelijk beschreven door Pallister in 1977. In de eerste publicaties rapporteerde deze onderzoeker twee gevallen van volwassen patiënten bij wie het beloop werd gekenmerkt door verschillende bevindingen: toevallen, musculaire hypotonie, intellectuele achterstand, musculoskeletale en organische misvormingen, ruwe gelaats- en huidskleur. veranderingen.
Tegelijkertijd beschreven Teschler-Nicola en Killiam in 1981 hetzelfde klinische beeld bij een driejarig meisje.
Daarom werd in de eerste klinische rapporten een algemene verwijzing gemaakt naar een medische aandoening die wordt gekenmerkt door de combinatie van aanvallen, verstandelijke beperking en een kenmerkend fysiek fenotype..
Bovendien was Gilgenkratz al in 1985 in staat om in het eerste geval tijdens de drachtfase iets te identificeren dat vandaag de dag gebruikelijk is dankzij moderne diagnostische technieken.
Pallister-Killiam-syndroom is een type genetische mozaïekziekte. In dit geval heeft de chromosomale verandering alleen invloed op enkele cellen van het lichaam. Er wordt een brede betrokkenheid van verschillende lichaamssystemen en organismen vastgesteld.
Het wordt voornamelijk gekenmerkt door een verstandelijke beperking, spierhypotonie, de ontwikkeling van onderscheidende gelaatstrekken, verandering van huidpigmentatie of haargroei, naast andere aangeboren veranderingen..
Bovendien is het Pallister-Kiliam-syndroom een zeldzame ziekte van aangeboren oorsprong die in de medische literatuur een groot aantal namen kan krijgen:
De prevalentiecijfers voor het Pallister-Killiam-syndroom zijn niet precies bekend. Er zijn niet veel definitieve diagnoses gesteld en de meeste hiervan zijn niet in de medische literatuur gepubliceerd.
Alle auteurs en instellingen definiëren dit syndroom dus als een zeldzame of niet vaak voorkomende genetische pathologie in de algemene bevolking..
Ongeveer 15 jaar geleden werd het Pallister-Killiam-syndroom in ongeveer 100 gevallen wereldwijd vastgesteld. Momenteel is dit cijfer meer dan 200 getroffen.
Epidemiologisch onderzoek heeft de incidentie van deze ziekte geschat op ongeveer 5,1 gevallen per miljoen pasgeboren kinderen, hoewel auteurs zoals Toledo-Bravo de la Laguna en medewerkers het op 1 / 25.000 schatten..
Een hogere prevalentie in verband met de sociodemografische kenmerken van de getroffenen is niet vastgesteld. Het Pallister-Killian-syndroom kan voorkomen in elk geslacht of technische en / of raciale groep.
Een breed scala aan tekenen en symptomen kan worden vastgesteld tijdens het klinische beloop van het Pallister-Killian-syndroom. Allemaal geassocieerd met craniofaciale en / of musculoskeletale afwijkingen en cognitieve veranderingen.
De ontwikkeling van cranio-faciale misvormingen vanaf de zwangerschapsfase tot de postnatale groei en de groei van het kind vormt een van de meest kenmerkende medische symptomen van het Pallister-Killiam-syndroom..
De meest voorkomende tekenen en symptomen zijn onder meer afwijkingen in de verschillende craniale en gezichtsstructuren die zullen leiden tot een ruw en atypisch uiterlijk:
Ondanks dat het minder significant is dan gezichtsveranderingen, is het heel gebruikelijk om verschillende musculoskeletale afwijkingen waar te nemen bij patiënten met het Pallister-syndroom:
Afwijkingen die verband houden met spierstructuur en mobiliteit zijn een andere van de belangrijkste klinische kenmerken van het Pallister-Killian-syndroom:
Spierhypotonie verwijst naar de identificatie van abnormaal verminderde spierspanning of spanning. Op visueel niveau kunnen slapheid en labiliteit worden waargenomen in verschillende spiergroepen, vooral geaccentueerd in de extremiteiten..
Aldus zal spier- en skeletpathologie een aanzienlijke vertraging veroorzaken in de verwerving van verschillende motorische vaardigheden, zowel in de neonatale periode als in de kindertijd..
Hoewel de ontwikkelingsperioden onder de getroffenen variabel zijn, bevat de meest voorkomende kalender de volgende mijlpalen:
Een ander sterk getroffen gebied is het zenuwstelsel. In de meeste gevallen houden de tekenen en symptomen voornamelijk verband met toevallen en verstandelijke beperkingen:
Hoewel ze minder vaak voorkomen, kunnen ook andere soorten medische complicaties optreden:
De oorsprong van het Pallister-Killian-syndroom wordt in verband gebracht met een genetische mozaïekafwijking op chromosoom 12. Het tast alleen het genetisch materiaal van sommige cellen in het lichaam aan..
Chromosomen maken deel uit van de kern van alle cellen die in het menselijk lichaam worden aangetroffen. Ze bestaan uit een grote verscheidenheid aan biochemische componenten en bevatten de genetische informatie van elk individu..
Mensen hebben 46 verschillende chromosomen, gerangschikt in paren en genummerd van 1 tot 23. Bovendien heeft elk chromosoom op individueel niveau een kort gebied of arm genaamd "p" en een lang genaamd "q"..
De afwijking beïnvloedt chromosoom 12 en resulteert in de aanwezigheid van een chromosoom met een abnormale structuur, een isochromosoom genaamd.
Dit chromosoom heeft dus de neiging om twee korte armen te hebben in plaats van één van elke p (korte) en lange (q) -configuratie..
Als gevolg hiervan zal de aanwezigheid van extra en / of abnormaal genetisch materiaal het normale en efficiënte verloop van de fysieke en cognitieve ontwikkeling van de getroffen persoon veranderen, wat aanleiding geeft tot de klinische kenmerken van het Pallister-Killian-syndroom..
Het Pallister-Killian-syndroom kan tijdens de zwangerschap of in de postnatale fase worden vastgesteld op basis van de klinische kenmerken en de resultaten van verschillende laboratoriumtests..
Tijdens de zwangerschap zijn de meest gebruikte tests echografie, vruchtwaterpunctie of vlokkentest. In die zin kan de analyse van het genetisch materiaal van het embryo ons een bevestiging van deze pathologie bieden door de identificatie van compatibele anomalieën..
Aan de andere kant, als de diagnose na de geboorte wordt gesteld, is het essentieel:
Er zijn geen specifieke therapieën ontwikkeld om mensen met het Pallister-Killian-syndroom te behandelen..
Dit syndroom wordt meestal geassocieerd met een slechte neurologische prognose en hoge sterftecijfers. Revalidatiebehandeling, speciaal onderwijs en ergotherapie kunnen echter een goede functionele prognose bieden en de kwaliteit van leven van de getroffenen verhogen..
Méndez en zijn werkteam (2013) beschrijven bijvoorbeeld een geval van revalidatiebehandeling die wordt gekenmerkt door:



Niemand heeft nog op dit artikel gereageerd.