
De Waardenburgsyndroom (SW) het is een pathologie van genetische oorsprong die wordt geclassificeerd als een soort neuropathie. De klinische kenmerken worden bepaald door de aanwezigheid van doofheid of gehoorverlies, abnormale pigmentatie van de ogen, het haar of de huid, en verschillende gezichtsveranderingen.
Deze pathologie wordt gekenmerkt door zijn brede symptomatische variabiliteit, daarom worden verschillende typen onderscheiden: Type I, Type II, Type III (Klein-Waardenburgsyndroom of psudo Waardenburg) en Type IV.

Op etiologisch niveau heeft het Waardenburg-syndroom een autosomaal dominant overervingspatroon. Het wordt meestal geassocieerd met specifieke mutaties in de genen EDN3, EDNRB, PAX3, SOX10, SNAI2 en MIT..
De diagnose wordt gesteld op basis van verschillende belangrijke en minder belangrijke klinische criteria. Het is echter noodzakelijk om verschillende aanvullende laboratoriumtests uit te voeren. Er is geen specifieke remedie of behandeling voor het Waardenburg-syndroom..
De interventie met deze pathologie richt zich meestal op de behandeling van gehoorstoornissen (chirurgische ingrepen, cochleaire implantaten, enz.), Logopedie en neuropsychologische revalidatie, evenals psychologische.
Artikel index
Dit syndroom werd aanvankelijk in 1848 beschreven door de Nederlandse geneticus en oogarts Petrus Johannes Waardenburg. In zijn klinische rapport verwees hij naar de belangrijkste klinische kenmerken:
Latere analyses identificeerden een grote klinische variabiliteit bij het Waardenbur-syndroom. Bovendien bracht Mckusick dit syndroom in verband met andere vergelijkbare klinische cursussen, zoals de ziekte van Hirschsprung..
Momenteel wordt het als een zeldzame pathologie beschouwd, die optreedt bij een variabele mate van gehoorbeschadiging die aanzienlijke veranderingen in het leren en de latere ontwikkeling van de getroffen persoon kan veroorzaken..
De prognose voor het Waardenburgsyndroom is gunstig, hoewel het geassocieerd kan zijn met significante morbiditeit en mortaliteit gerelateerd aan medische complicaties, vooral intestinale complicaties..
Het Waardenburg-syndroom is een aangeboren genetische aandoening waarvan de tekenen en symptomen sterk variëren tussen de getroffenen..
De meest voorkomende kenmerken zijn onder meer opvallende gezichtsafwijkingen, veranderde pigmentatie van de huid, ogen of haar en doofheid..
In de medische literatuur wordt dit syndroom vaak beschouwd als een vorm van genodermatose of neuropathie. De term genodermatose verwijst naar een brede reeks ziekten die worden gekenmerkt door de aanwezigheid van huidafwijkingen en veranderingen van genetische oorsprong..
Aan de andere kant verwijst de term neuropathie naar een groep pathologieën die zijn afgeleid van de ontwikkeling van anomalieën en defecte processen tijdens de migratie en differentiatie van de cellen van de neurale top tijdens de zwangerschap..
De neurale top is een embryonale structuur die bestaat uit een brede reeks ongedifferentieerde cellen waarvan de ontwikkeling zal leiden tot de vorming van de cranio-faciale structuur en de neuronale en gliacellen die een groot deel van het zenuwstelsel zullen vormen..
Tussen de 8e en 10e week van de zwangerschap begint meestal het migratieproces van de cellen waaruit de neurale top bestaat. Wanneer verschillende pathologische factoren of genetische afwijkingen dit proces verstoren, kunnen significante cognitieve en / of fysieke afwijkingen optreden, zoals het Waardenburgsyndroom..
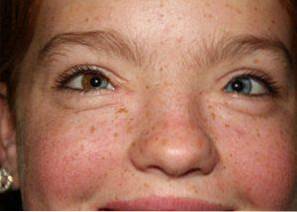
De prevalentie van het Waardenbur-syndroom wordt geschat op 1 geval op elke 40.000 mensen wereldwijd. Sinds de ontdekking ervan zijn ongeveer 1.400 verschillende gevallen beschreven in de medische en experimentele literatuur..
Het lijkt mannen en vrouwen in gelijke mate te beïnvloeden. Er zijn geen associaties met geografische regio's of bepaalde etnische en raciale groepen geïdentificeerd.
Het Waardenbug-syndroom vertegenwoordigt 2-5% van alle gediagnosticeerde gevallen van aangeboren gehoorverlies.
Hoewel er verschillende klinische cursussen zijn geïdentificeerd, komen type I en II het meest voor. Type III en IV zijn zeldzaam.

Het Waardenburg-syndroom wordt gekenmerkt door drie fundamentele veranderingen: craniofaciale veranderingen, pigmentafwijkingen en doofheid:
Een andere centrale medische bevindingen van het Waardenburg-syndroom is het verlies van gehoorvermogen en scherpte. De meest voorkomende is om bij getroffenen een variabele mate van doofheid of perceptief gehoorverlies te identificeren.
De term perceptief gehoorverlies verwijst naar een verlies van gehoorvermogen als gevolg van interne verwondingen die verband houden met de zenuwuiteinden die auditieve informatie van het binnenoor naar de hersencentra leiden..

Het Waardenburg-syndroom wordt ingedeeld in 4 basistypen op basis van het klinische beloop en de specifieke symptomen die aanwezig zijn bij getroffen mensen:
Het Waardenbuug-syndroom heeft een aangeboren oorsprong die verband houdt met verschillende genetische veranderingen.
De analyse van gevallen heeft het mogelijk gemaakt om deze anomalieën in de genen te lokaliseren: EDN3, EDNRB, PAX3, SOX10, SNAI2 en MIT.
Deze set genen lijkt betrokken te zijn bij de ontwikkeling en vorming van verschillende soorten cellen, waaronder de cellen die verantwoordelijk zijn voor de productie van melanocyten..
Melanocyten zijn verantwoordelijk voor het aanmaken van melanine, een pigment dat bijdraagt aan de verkleuring van de ogen, het haar of de huid.
Afhankelijk van de verschillende klinische cursussen, kunnen we verschillende genetische veranderingen identificeren:
Zoals we in de eerste beschrijving hebben opgemerkt, wordt de diagnose van het Waardenbug-syndroom gesteld op basis van verschillende belangrijke en minder belangrijke criteria:
Om een definitieve diagnose te stellen, is het essentieel om de aanwezigheid van twee hoofdcriteria of ten minste één hoofdcriterium en twee minderjarig criterium vast te stellen. Bovendien is het nodig om enkele aanvullende tests te gebruiken: biopsie, audiometrie of genetische tests.
Er is geen remedie voor het Waardenbug-syndroom, hoewel symptomatische benaderingen kunnen worden gebruikt..
Behandeling van de meest voorkomende tekenen en symptomen vereist meestal medische tussenkomst van dermatologen en oogartsen..
Aan de andere kant kan in het geval van de behandeling van perceptieve doofheid een cochleair implantaat worden uitgevoerd in combinatie met een logopedie en neuropsychologische interventie..



Niemand heeft nog op dit artikel gereageerd.