
De Williams-syndroom het is een ontwikkelingsstoornis van genetische oorsprong die wordt geassocieerd met een karakteristiek profiel van fysieke en cognitieve stoornissen. Specifiek op klinisch niveau wordt het gekenmerkt door 4 hoofdpunten: 1) atypische gelaatstrekken en kenmerken, 2) gegeneraliseerde vertraging in psychomotorische ontwikkeling en specifiek neurocognitief profiel, 3) cardiovasculaire veranderingen en t) mogelijkheid om infantiele hypercalciëmie te ontwikkelen.
Ondanks het feit dat het Williams-syndroom als een zeldzame ziekte wordt beschouwd, zijn er duizenden getroffen mensen over de hele wereld. Met betrekking tot de diagnose levert het klinische onderzoek meestal de nodige bevindingen op voor de vaststelling ervan, maar om andere pathologieën en fout-positieven uit te sluiten, wordt een genetisch onderzoek meestal gestart met behulp van verschillende technieken..
Aan de andere kant is er geen remedie voor het Williams-syndroom, noch een standaard behandelingsprotocol, dus de meeste therapeutische interventies zullen proberen medische complicaties te reguleren. Daarnaast is het essentieel om vroege zorgprogramma's, geïndividualiseerd speciaal onderwijs en neuropsychologische stimulatie in de interventies op te nemen..
Artikel index
Williams-syndroom is een ontwikkelingsstoornis die verschillende gebieden aanzienlijk kan beïnvloeden.
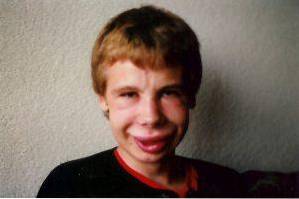
Over het algemeen wordt deze pathologie gekenmerkt door de aanwezigheid van atypische gelaatstrekken of cardiovasculaire veranderingen, matige verstandelijke beperking, leerproblemen en onderscheidende persoonlijkheidskenmerken..
Zo werd de eerste patiënt met het Williams-syndroom beschreven door Dr. Guido Fanconi in een klinisch rapport uit 1952. Het was echter de cardioloog Joseph Williams die in 1961 deze pathologie nauwkeurig identificeerde, en die ook werd beschreven door de Duitse Beuren..
Hierdoor krijgt het Williams-syndroom zijn naam van beide auteurs (Williams-Beuren-syndroom), of gewoon van de eerste.
Ondanks het feit dat tot een paar jaar geleden de identificatie van de pathologie werd uitgevoerd op basis van de fenotypische kenmerken, vonden Edward et al. In 1993 een genetische afwijking in chromosoom 7q 11.23 als etiologische oorzaak..
Hoewel het Williams-syndroom wordt geassocieerd met een breed scala aan secundaire medische complicaties, heeft het geen hoog sterftecijfer. In veel gevallen kunnen getroffen personen een onafhankelijk functioneel niveau bereiken.
Het Williams-syndroom wordt als een zeldzame of zeldzame genetische aandoening beschouwd.
De Williams Syndrome Association, en andere instellingen, hebben geschat dat het Williams syndroom een prevalentie heeft van ongeveer 1 geval per 10.000 mensen wereldwijd. Specifiek is vastgesteld dat er in de Verenigde Staten ongeveer 20.000 of 30.000 slachtoffers kunnen zijn.
Met betrekking tot de verdeling van de pathologie naar geslacht zijn er geen recente gegevens die wijzen op een hogere prevalentie in een van hen, bovendien zijn er geen verschillen vastgesteld tussen geografische regio's of etnische groepen..
Aan de andere kant weten we ook dat het Williams-syndroom een sporadische medische aandoening is, hoewel er enkele gevallen van familieoverdracht zijn beschreven..
Het Williams-syndroom heeft, net als andere pathologieën van genetische oorsprong, een klinisch beloop dat wordt gekenmerkt door multisysteembetrokkenheid.
Veel auteurs, zoals González Fernández en Uyaguari Quezada, beschrijven het klinische spectrum van het Williams-syndroom onderverdeeld in verschillende gebieden: biomedische kenmerken, psychomotorische en cognitieve kenmerken, psychologische en gedragskenmerken, onder anderen..
De fysieke aandoening die aanwezig is bij het Wiliams-syndroom is divers, een van de meest voorkomende klinische bevindingen die we kunnen waarnemen:
Een vertraagde of vertraagde ontwikkeling kan al tijdens de zwangerschap worden vastgesteld. Kinderen met het Williams-syndroom worden vaak geboren met een laag gewicht en een lage lengte. Bovendien is, zodra het volwassen stadium is bereikt, de totale lengte meestal lager dan die van de algemene bevolking, ongeveer 10-15 cm.
Gezichtsveranderingen zijn een van de meest kenmerkende klinische bevindingen bij dit syndroom. Bij getroffen personen kunnen we een aanzienlijk smal voorhoofd, duidelijke huidplooien in de oogspleet, scheelzien, stervormige iris, korte en afgeplatte neus, prominente jukbeenderen en een kleinere kin dan normaal zien..
In het geval van veranderingen die verband houden met de ontwikkeling van spieren en botten, is het mogelijk om de aanwezigheid van verminderde spierspanning en kracht, gewrichtslaxiteit, scoliose, contracturen, onder andere waar te nemen. Visueel kan een houding worden waargenomen die wordt gekenmerkt door afhangende schouders en half gebogen onderste ledematen..
Hoewel er meestal geen significante afwijkingen of misvormingen in de gehoorschelp zijn, ontwikkelt zich in alle gevallen een toename van de gehoorgevoeligheid. Getroffen personen hebben de neiging om bepaalde geluiden als vervelend of pijnlijk waar te nemen of te ervaren.
De huid heeft meestal weinig elasticiteit, dus het is mogelijk om vroege tekenen van veroudering waar te nemen. Bovendien kunnen hernia's ontstaan, vooral in de lies- en navelstreek..
De verschillende afwijkingen in het hart en de bloedvaten vormen de belangrijkste medische complicatie, aangezien ze het voortbestaan van de getroffen persoon in gevaar kunnen brengen.
Onder de cardiovasculaire anomalieën zijn enkele van de meest voorkomende supravalvulaire aortastenose, pulmonale takstenose, aortaklepstenose. Al deze veranderingen, op klinisch niveau, kunnen andere vasculaire gebieden en zelfs de hersenen beïnvloeden, vanwege de ontwikkeling van arteriële hypertensie.
Afwijkingen die verband houden met de nierfunctie en de blaas komen zeer vaak voor. Bovendien kan ook een ophoping van calcium (nefrocalcinose), urinaire urgentie of nachtelijke enuresis worden gedetecteerd.
Op cognitief niveau worden de belangrijkste kenmerken gevormd door een algemene vertraging in de verwerving van motorische vaardigheden, een matige intellectuele achterstand en verschillende veranderingen die verband houden met de visuele waarneming..
Er worden verschillende veranderingen beschreven die verband houden met evenwichts- en coördinatieproblemen, die voornamelijk te wijten zijn aan de aanwezigheid van musculoskeletale afwijkingen en die onder meer een vertraging in gangverwerving, eindmotoriek, enz. Zullen veroorzaken..
Het is mogelijk om een matige mentale retardatie te vinden, het typische IQ van de getroffenen schommelt meestal tussen 60 en 70. Met betrekking tot de specifieke getroffen gebieden is er een duidelijke asymmetrie: naast psychomotorische coördinatie, perceptie en visuele integratie, is het meestal worden duidelijk beïnvloed, terwijl gebieden zoals taal meestal meer ontwikkeld zijn.
In de meest beginstadia is er meestal een vertraging bij het verwerven van taalvaardigheden, maar dit herstelt zich meestal na ongeveer 3-4 jaar. Kinderen met het Williams-syndroom hebben de neiging om een goede expressieve communicatie te hebben, zijn in staat om gecontextualiseerde woordenschat te gebruiken, de grammatica te corrigeren, oogcontact, gezichtsuitdrukkingen, enz..
Een van de belangrijkste bevindingen bij het Williams-syndroom is het uitzonderlijke sociale gedrag van de getroffenen. Hoewel in sommige gevallen angstcrises of overmatige zorgen kunnen voorkomen, zijn ze erg empathisch en gevoelig.
Het meest recente onderzoek heeft aangetoond dat de oorzaak van het Williams-syndroom wordt gevonden in verschillende genetische veranderingen op chromosoom 7. Chromosomen bevatten de genetische informatie van elke persoon en bevinden zich in de kern van de lichaamscellen..
Bij mensen kunnen we 46 chromosomen vinden die in paren zijn verdeeld. Deze zijn genummerd van 1 tot 23, behalve het laatste paar dat bestaat uit de geslachtschromosomen, XX genoemd in het geval van vrouwen, XY in het geval van mannen. Binnen elk chromosoom kan er dus een oneindig aantal genen zijn.
In het bijzonder is het abnormale proces dat wordt geïdentificeerd bij het Williams-syndroom een microkeuze of afbraak van een DNA-molecuul dat dit chromosoom bevestigt. Normaal gesproken vindt dit type fout plaats in de ontwikkelingsfase van de mannelijke of vrouwelijke gameten..
Genetische afwijkingen worden gevonden in het 7q11.23-gebied, waarin meer dan 25 verschillende genen zijn geïdentificeerd die verband houden met het kenmerkende klinische patroon van deze pathologie..
Sommige van de genen, zoals Clip2, ELN, GTF21, GTF2IRD1 of LIMK1, zijn afwezig bij de getroffenen. Verlies van ELN is gerelateerd aan bindweefsel, huid en cardiovasculaire afwijkingen.
Anderzijds geeft enig onderzoek aan dat het verlies van de Clip2-, GTF2I-, GTF2IRD1- en LIMK1-genen veranderingen in visuoperceptieve processen, gedragsfenotype of cognitieve gebreken kan verklaren..
Bovendien lijkt specifiek het GTF2IRD1-gen een prominente rol te spelen bij de ontwikkeling van atypische gelaatstrekken. Van zijn kant lijkt het NCF1-gen verband te houden met een hoog risico op het ontwikkelen van hypertensie.
Tot voor kort werd de diagnose van het Williams-syndroom uitsluitend gesteld op basis van de observatie van fenotypische kenmerken (gezichtsafwijkingen, verstandelijke beperking, specifieke cognitieve gebreken, onder andere).
Momenteel wordt de diagnose van het Williams-syndroom echter meestal in twee fasen gesteld: analyse van de klinische bevindingen en bevestigende genetische studies. De klinische diagnose omvat dus meestal:
- Lichamelijk en neurologisch onderzoek en evaluatie.
- Analyse van groeiparameters.
- Cardiorespiratoir systeemonderzoek.
- Nefrourologisch onderzoek.
- Analyse van calciumspiegels in urine en bloed.
- Oogheelkundige analyse.
Aan de andere kant wordt genetische analyse gebruikt om de aanwezigheid van genetische veranderingen die compatibel zijn met het Williams-syndroom te bevestigen, een van de meest gebruikelijke tests is de fluorescente in situ hybridisatie (FIHS) -techniek..
Na de extractie van een bloedmonster wordt de in situ hybridisatietechniek uitgevoerd door DNA-sondes te markeren die onder fluorescerend licht worden gedetecteerd..
Er is geen specifieke behandeling voor het Williams-syndroom, maar deze pathologie gaat gepaard met meerdere complicaties in verschillende organen, dus medische interventies zullen gericht zijn op de behandeling ervan..
De auteurs González Fernández en Uyaguari Quezada benadrukken dat alle interventies een uitgesproken multidisciplinair karakter moeten hebben, zodat de symptomatische variëteit die kenmerkend is voor dit syndroom, kan worden behandeld. Daarnaast wijzen ze ook op verschillende therapeutische maatregelen, afhankelijk van het getroffen gebied:
In dit geval vereisen medische complicaties zoals hartveranderingen of musculoskeletale misvormingen meestal een behandeling die voornamelijk gebaseerd is op de toediening van medicijnen en chirurgische ingrepen. Medische professionals uit verschillende gebieden (kinderartsen, cardiologen, oogartsen, enz.) Nemen gewoonlijk deel aan de behandeling van lichamelijke symptomen..
Cognitieve gebreken zoals visus-perceptuele verandering of taalachterstand moeten vanaf de vroege stadia worden aangepakt. Cognitieve stimulatie en revalidatie zullen een bepalende factor zijn bij het bereiken van een autonoom leven op volwassen leeftijd.
Hoewel degenen die getroffen zijn door het Williams-syndroom meestal een goed sociaal functioneren vertonen, vertonen ze in sommige gevallen de neiging om buitengewoon angstig gedrag te vertonen en volhardend gedrag of fobieën te ontwikkelen..
Daarom is het in deze gevallen essentieel om een psychologische benadering te implementeren, door middel van verschillende strategieën die effectief zijn om deze problemen of moeilijkheden te minimaliseren..



Niemand heeft nog op dit artikel gereageerd.